Harnessing hidden genetic information in clinical data with REGLE
July 18, 2024
Taedong Yun, Software Engineer, and Farhad Hormozdiari, Research Scientist, Google Research
Quick links
Modern healthcare systems generate a vast amount of high-dimensional clinical data (HDCD), such as spirogram measurements, photoplethysmograms (PPG), electrocardiogram (ECG) recordings, CT scans, and MRI imaging, that cannot be summarized as a single binary or a continuous number (cf. "has asthma" or "height in centimeters"). Understanding the connection between our genomes and HDCD not only improves our understanding of diseases but is also crucial to the development of disease treatments.
HDCH are stored in electronic health records and large biobank projects, such as UK Biobank in the United Kingdom, BioBank Japan in Japan, and All of Us in the United States. These projects obtain participant consent before de-identifying data and sharing a portion of this valuable resource with qualified scientists. The goal is to enhance the prevention, diagnosis, and treatment of various life-threatening illnesses.
The genomics team at Google Research has made progress utilizing HDCD for characterizing diseases or biological traits like optic nerve head morphology and chronic obstructive pulmonary disease (COPD). In an effort to better understand the genetic architecture of these particular traits, we previously performed genome-wide association studies (GWAS) on the trait predictions generated by supervised machine learning (ML) models. However, obtaining large enough volumes of data that contain disease labels to train supervised ML models is not always possible. Furthermore, simple disease labels cannot fully capture the biology embedded in the underlying data, and we lack statistical methods to directly utilize HDCD in genetic analysis like GWAS.
To overcome these limitations, in "Unsupervised representation learning on high-dimensional clinical data improves genomic discovery and prediction", published in Nature Genetics, we introduce a principled method to study the underlying genetic contributors to the general organ functions that are reflected in the HDCD. REpresentation learning for Genetic discovery on Low-dimensional Embeddings (REGLE) is a computationally efficient method that requires no disease labels, and can incorporate information from expert-defined features (EDFs) when they are available.
Uncovering hidden information in HDCD
A simple approach to study the connection between genes and HDCD is to perform GWAS on each data coordinate, e.g., one can study the variations in each pixel value in medical images. This approach is computationally expensive and has low power to discover significant associations due to the high correlation between nearby coordinates and the massive multiple-testing burden. A more widely-used approach is to focus on a small number of expert-defined features (EDFs) extracted from the HDCD as the target traits or phenotypes of the GWAS. EDFs can include clinically known features, such as forced vital capacity (FVC) or forced expiratory volume in 1 second (FEV1) in the case of spirograms, as illustrated in previous work. While these EDFs are important features discovered by subject matter experts, we hypothesized that they may not comprehensively capture the signals encoded in HDCD, and thus running GWAS on these may not exploit the full potential of HDCD.
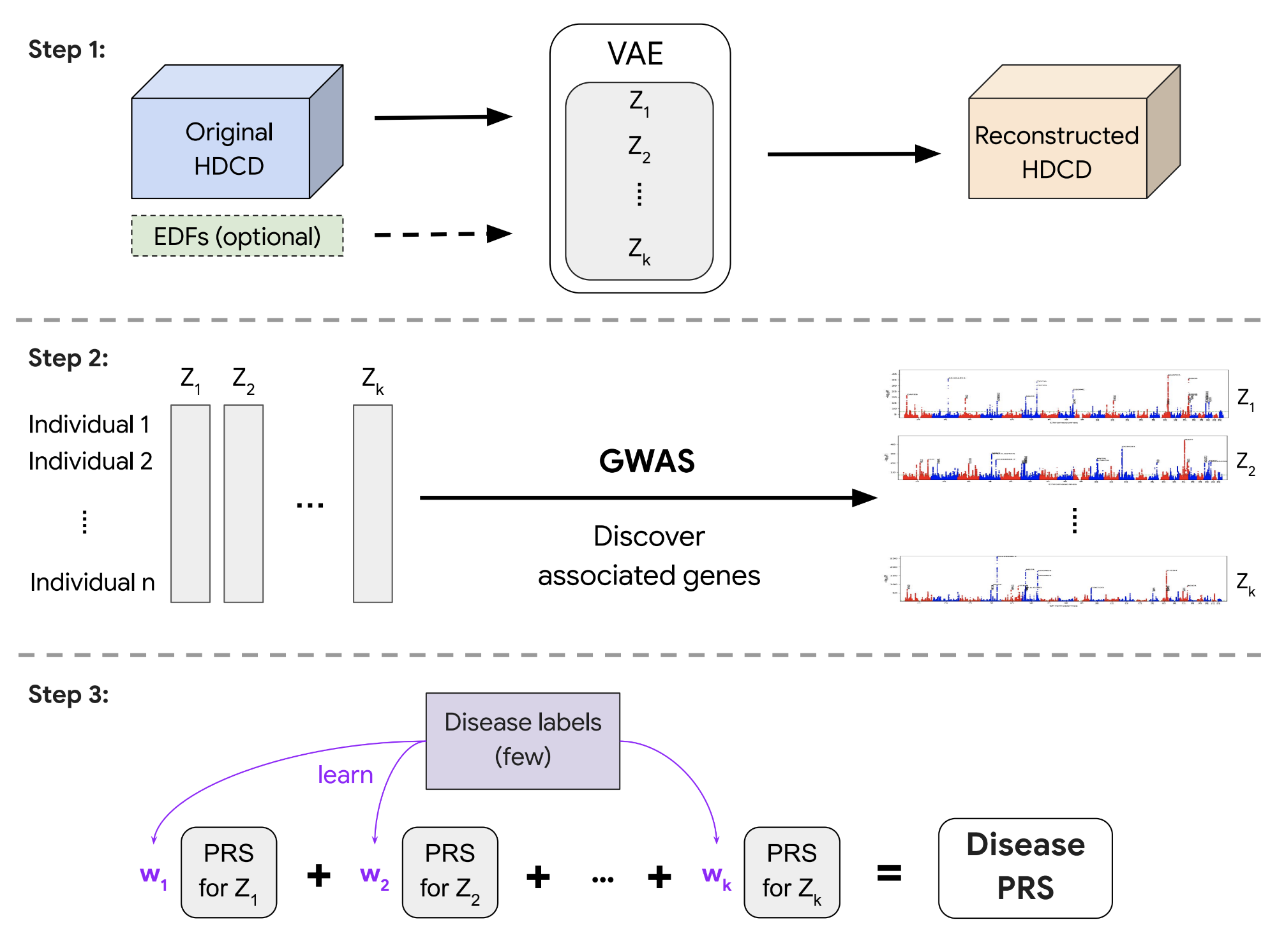
REGLE aims to overcome these limitations using a variational autoencoder (VAE) model. The method consists of three main steps: (1) learning a non-linear, low-dimensional, disentangled representation (i.e., encodings or embeddings) of the HDCD via VAE; (2) performing GWAS on each encoding coordinate; and (3) using polygenic risk scores (PRSs) from the encoding coordinates as genetic scores of general biological functions and then potentially combining these scores to create a PRS for a specific disease or trait (given a small number of disease labels). Notably, REGLE also enables relevant EDFs to be optionally included in the input to the decoder in a modified VAE architecture, so that the encoder is encouraged to learn only the residual signals not represented by the EDFs.
REGLE consists of three steps: 1) learning the lower-dimensional embeddings, 2) performing GWAS on each coordinate of the embeddings, and 3) creating a polygenic disease risk score while requiring only a small number of disease labels.
Detecting novel genetic loci for lung and circulatory functions
We demonstrate the capabilities of REGLE using two high-dimensional clinical data modalities: spirograms that measure lung function and PPGs that measure cardiovascular function. Both can be collected in a noninvasive, relatively inexpensive way in clinics or from consumer wearable devices, and there are well-known features for both modalities (e.g., FEV1 or FVC for spirograms and presence or location of a dicrotic notch for PPGs). Compared to genome-wide association studies on spirogram and PPG features with the same dimension, REGLE’s studies on the learned encodings recover most known genetic loci linked to lung and circulatory function while also detecting additional loci (e.g., 45% more significant loci for PPG). If these loci are validated in further analysis and wet-lab experiments, they have a potential to become novel drug targets.
Improved genetic risk scores
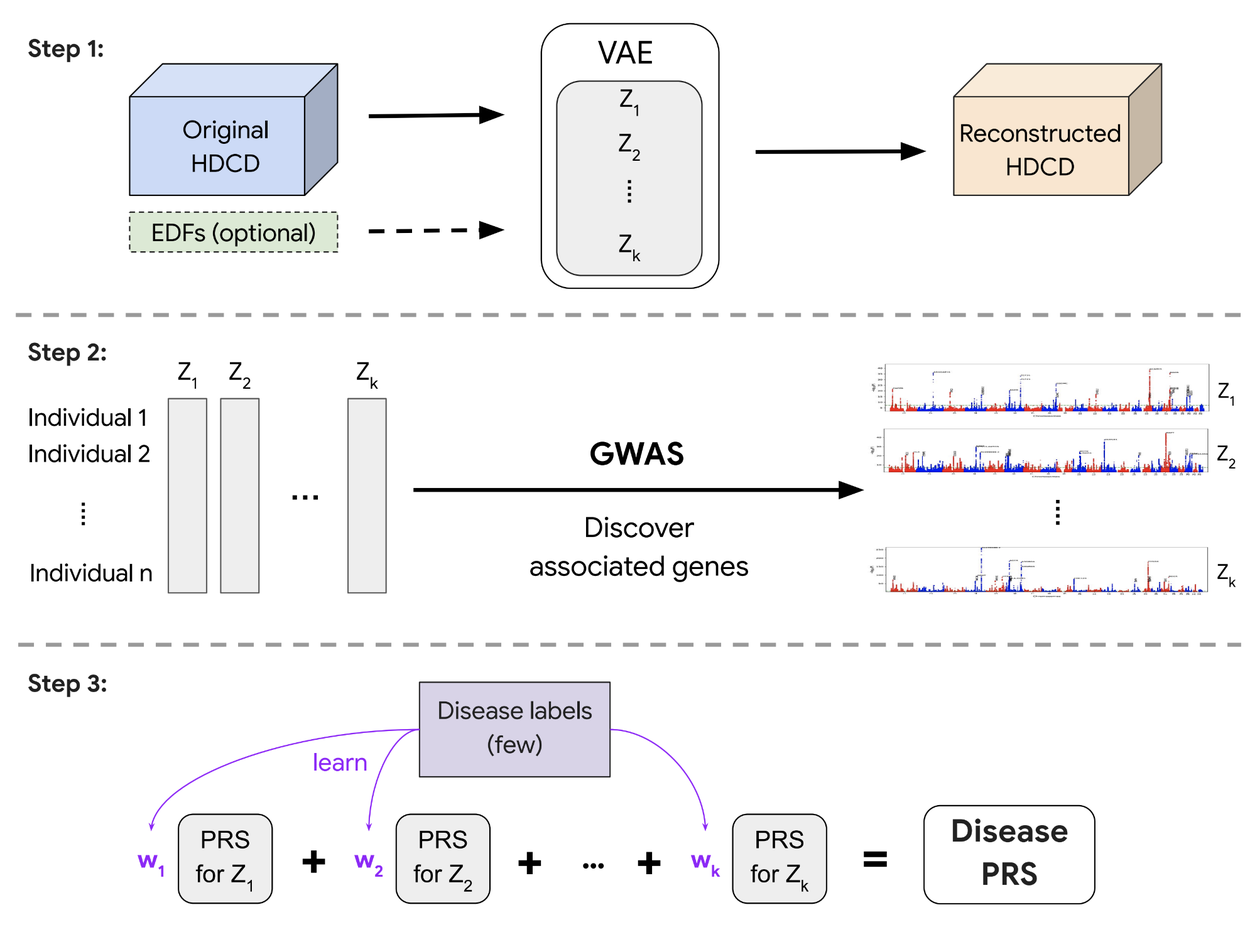
A polygenic risk score (PRS) is a summary of estimated effects of many genetic variants on a particular trait, represented as a single number. PRSs created by genome-wide association studies on REGLE embeddings can be combined using only a small number of disease labels to generate a PRS for that particular disease (Step 3 above). We observed that the lung function PRSs created from spirogram encodings improved COPD and asthma predictions compared to existing methods (such as PRSs generated by expert-defined features, PCA, and spline fitting), and stratified the risk groups more effectively than the feature PRS on both ends of the risk spectrum. We observed statistically significant improvement in multiple metrics (AUC-ROC, AUC-PR, and Pearson correlation) in multiple independent datasets (COPDGene, eMERGE III, Indiana Biobank, and EPIC-Norfolk) for asthma and COPD, as shown below.
Comparison of spirogram encodings (SPINC) and residual spirogram encodings (RSPINC) PRSs with expert-defined feature PRSs on asthma prevalence. The horizontal dashed line shows the total prevalence. Lower is better for the bottom percentiles; Higher is better for the top percentiles. * indicates a statistically significant difference.
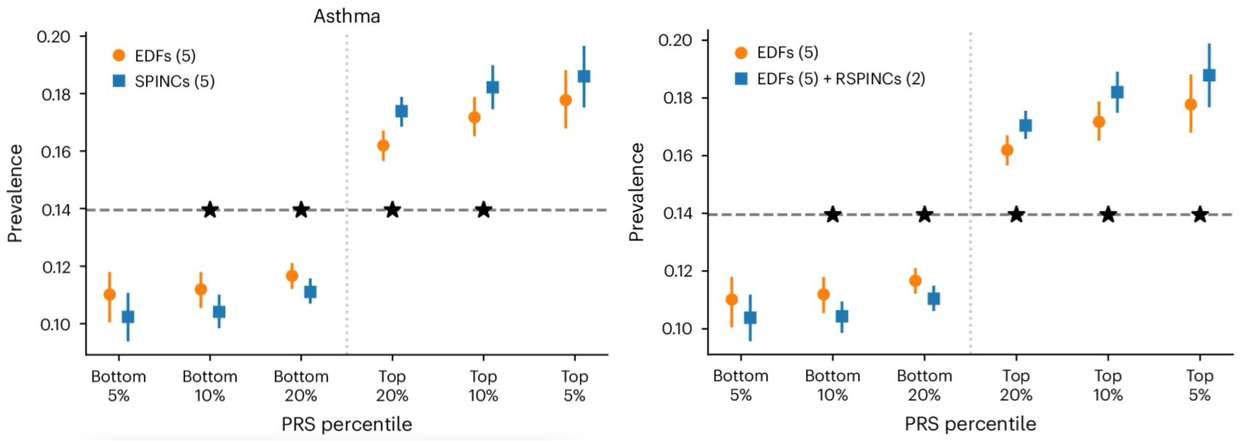
Similarly, PRS derived from REGLE embeddings of PPG improve hypertension and systolic blood pressure (SBP) predictions. We worked with academic collaborators to evaluate hypertension and SBP PRS generated by PPG encodings and PPG features in three independent datasets (COPDGene, eMERGE III, and EPIC-Norfolk) in addition to the held-out test set from the UK Biobank. We observed a consistent trend of improvement from using PRSs from PPG encodings over those from expert-defined features, both for hypertension and SBP in multiple datasets.

Comparison of PPG encoding (PLENC) PRSs for systolic blood pressure (SBP). SBP is a quantitative trait so Pearson & Spearman correlations are computed. * indicates a statistically significant difference.
Partially interpretable embeddings
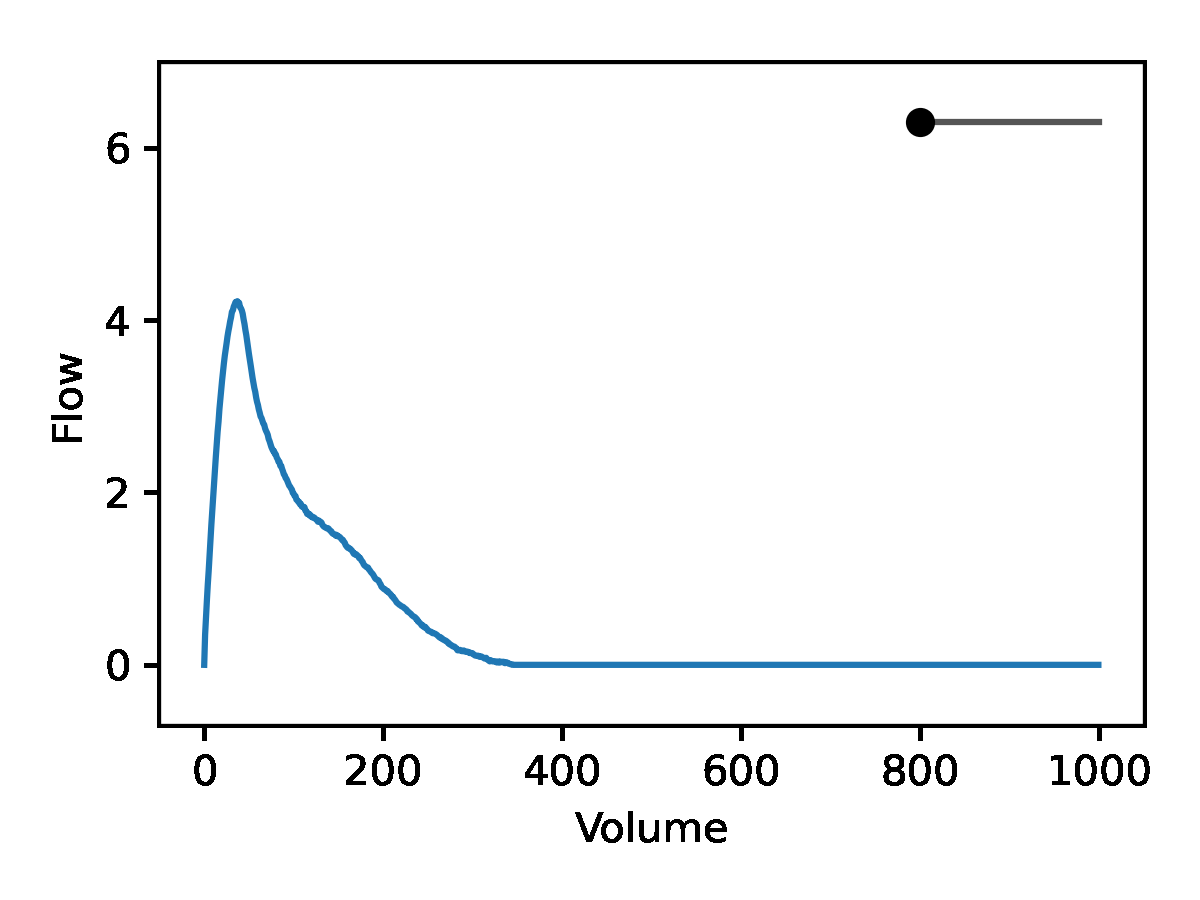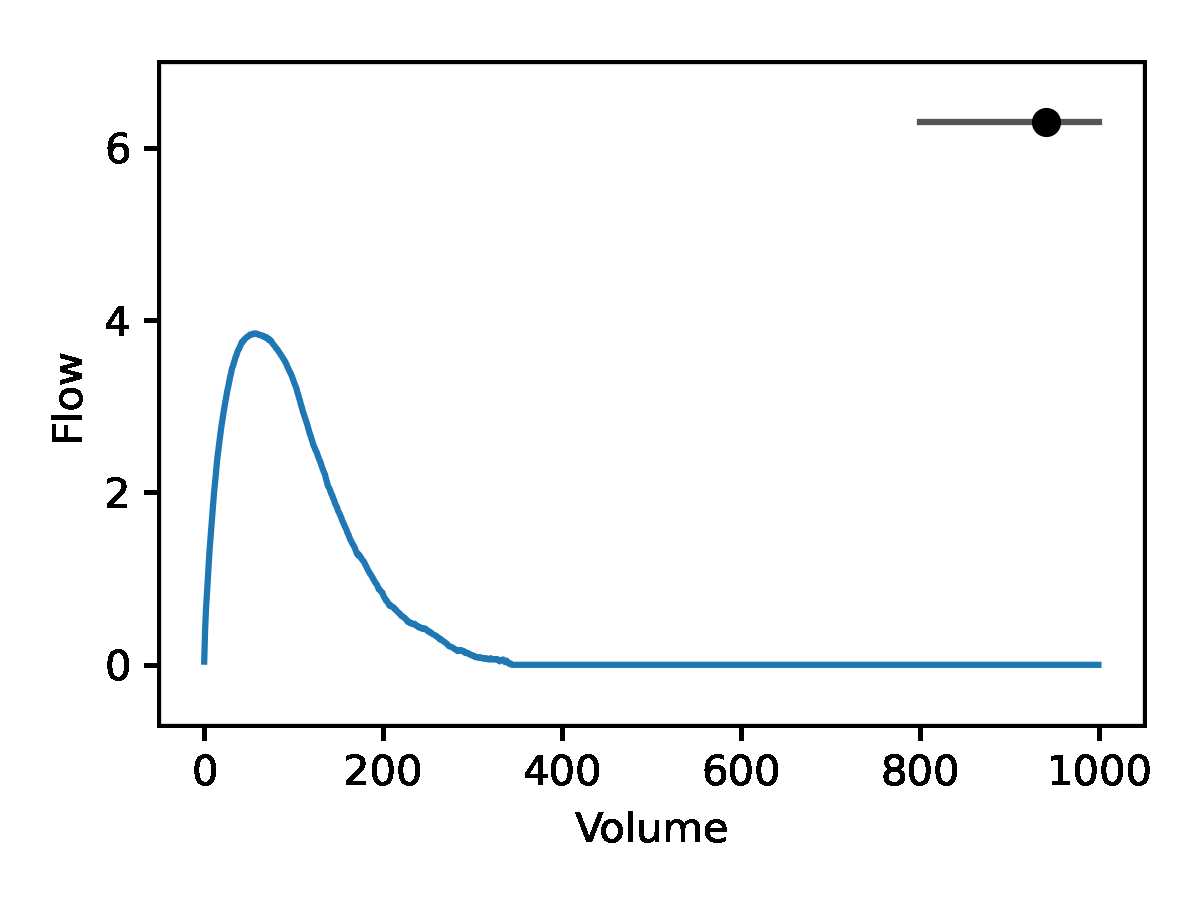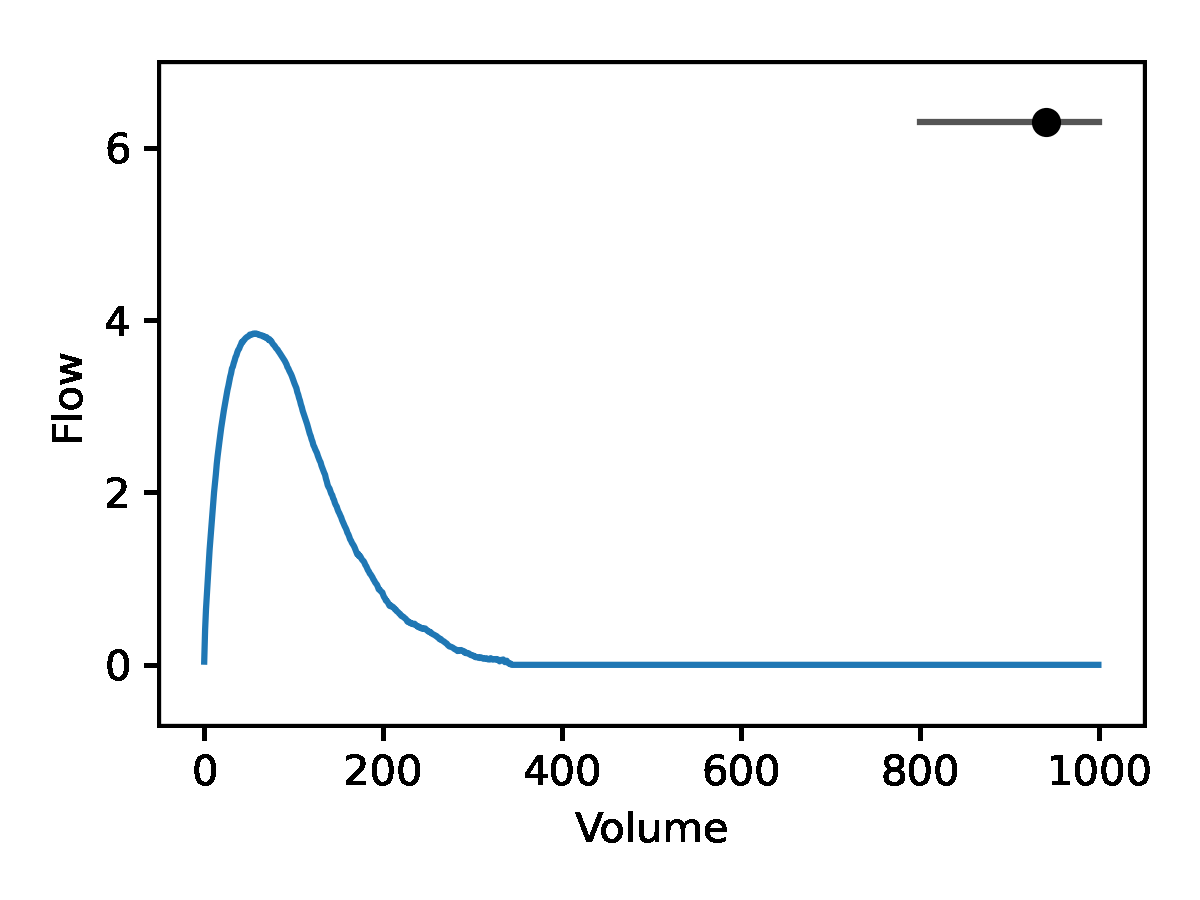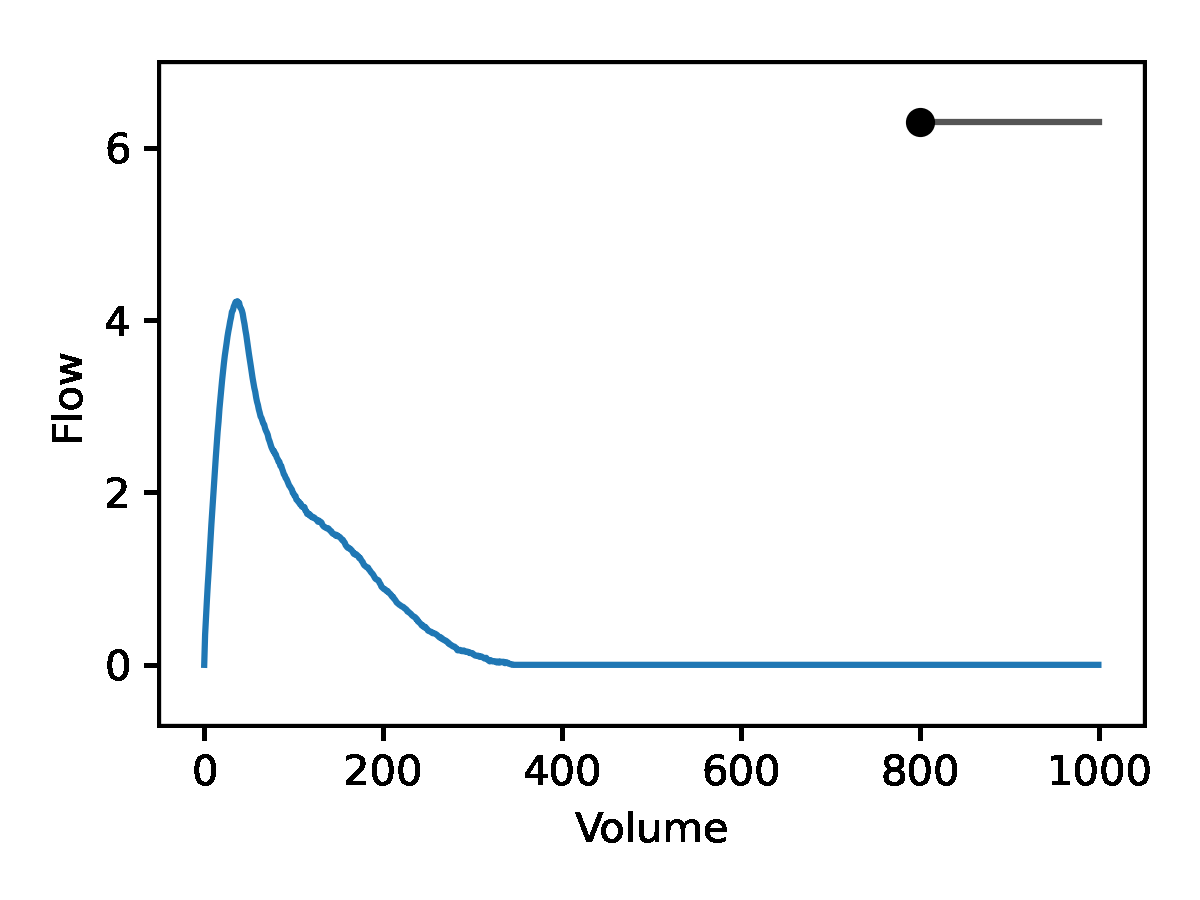
Leveraging the generative nature of REGLE, we studied the influence of encoding coordinates on spirogram shape by fixing the values of expert-defined features and varying one encoding coordinate while keeping the other ones at zero. We then generate the corresponding spirograms using only the decoder portion of the trained model. A typical flow-volume spirogram consists of two distinct parts: (1) a relatively brief part to reach peak flow, where the flow increases monotonically as the volume increases, and (2) the main part of the spirogram, where the flow decreases monotonically. The figure below shows that varying the first coordinate amounts to widening or narrowing of the second part (negative slope) while keeping the first part relatively fixed. In fact, the concavity of the second part of the curve is well-known to pulmonologists as coving, an indicator of airway obstruction that is not well-represented by the standard EDFs.
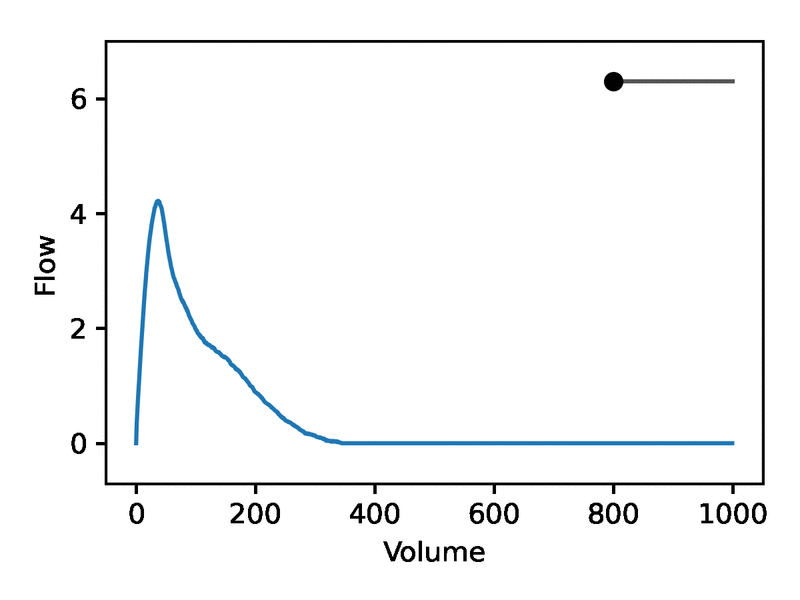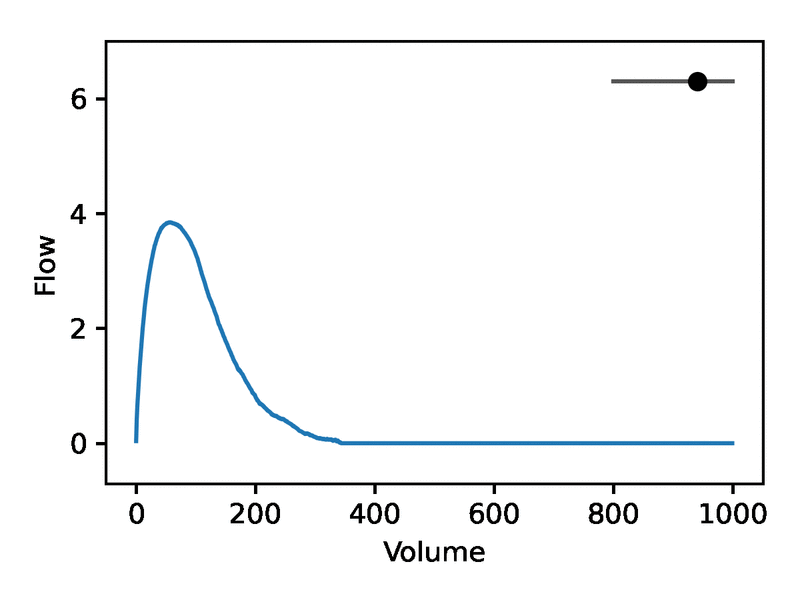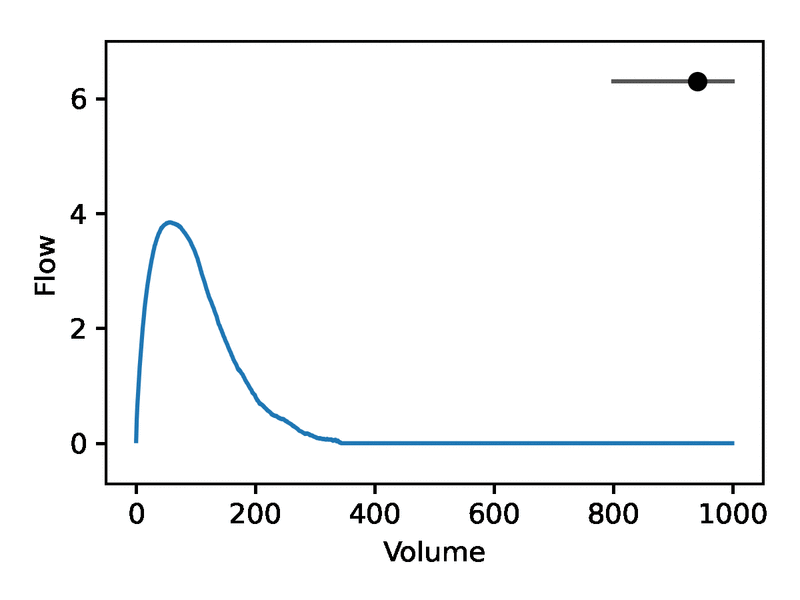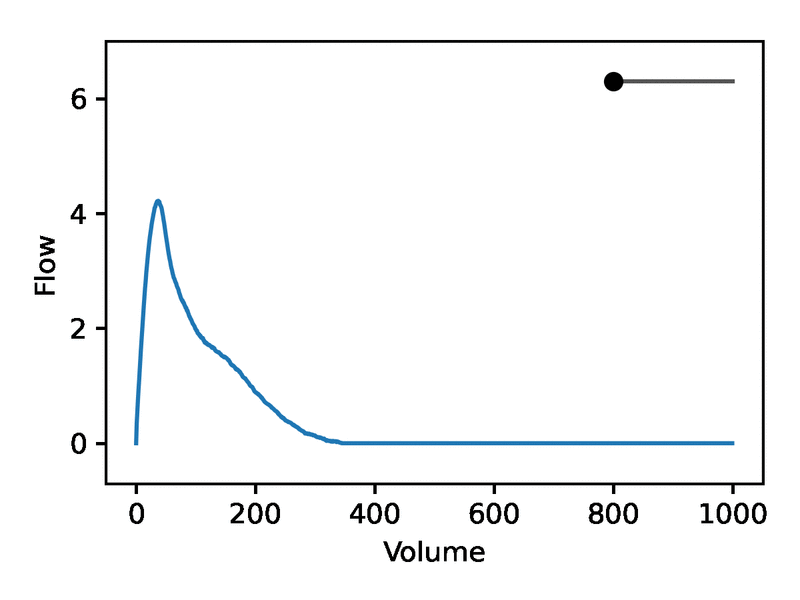
The effect of varying one spirogram encoding coordinate.
Conclusion
We presented REGLE, an unsupervised learning method that performs genetic analysis, improved novel loci discovery, and risk prediction to overcome limitations from our previous work on ML-based phenotyping. Unsupervised learning of HDCD representations for genomic discovery is attractive owing to the difficulty of manually discovering EDFs at scale. The REGLE framework also supports the principled use of such features in modeling by modifying the traditional VAE architecture. We demonstrate REGLE in two clinical data modalities, spirograms and PPGs, which can be routinely measured in clinical settings and can also be measured passively and noninvasively via smartphones or wearable devices.
REGLE provides a mechanism for identifying genetic influences on organ function in the absence of labeled data and naturally admits to incorporating expert features into the model. It also provides a method to create disease- and trait-specific PRS with very few labels. As biobanks with rich imaging, activity monitoring, medical records and paired genetic data continue to grow, we anticipate that this or similar methods will be increasingly used to further elucidate the genetic underpinnings of human traits and diseases.
Acknowledgments
This work is the combined output of multiple contributors and institutions. We thank all collaborators: Justin Cosentino, Babak Behsaz, Yuchen Zhou, Zachary R. McCaw, Howard Yang, Andrew Carroll, Cory Y. McLean (Google), Davin Hill (Northeastern University), Tae-Hwi Schwantes-An, Dongbing Lai (Indiana University), John Bates (Verily), Brian D. Hobbs, Michael H. Cho (Brigham and Women’s Hospital & Harvard Medical School), Robert Luben, Anthony P. Khawaja (Moorfields Eye Hospital & University College London). We also thank Nick Furlotte for reviewing the manuscript, Greg Corrado and Shravya Shetty for support, and Annisah Um'rani for helping with publication logistics.
Quick links
Other posts of interest
-

March 31, 2026
Building better AI benchmarks: How many raters are enough?- Algorithms & Theory ·
- Machine Intelligence
-

March 25, 2026
Vibe Coding XR: Accelerating AI + XR prototyping with XR Blocks and Gemini- Human-Computer Interaction and Visualization ·
- Machine Intelligence
-

March 24, 2026
TurboQuant: Redefining AI efficiency with extreme compression- Algorithms & Theory ·
- Generative AI ·
- Machine Intelligence

Comparison of spirogram encodings (SPINC) and residual spirogram encodings (RSPINC) PRSs with expert-defined feature PRSs on asthma prevalence. The horizontal dashed line shows the total prevalence. Lower is better for the bottom percentiles; Higher is better for the top percentiles. * indicates a statistically significant difference.