Unlocking the "Chemome" with DNA-Encoded Chemistry and Machine Learning
June 11, 2020
Posted by Patrick Riley, Principal Engineer, Accelerated Science Team, Google Research
Quick links
Much of the development of therapeutics for human disease is built around understanding and modulating the function of proteins, which are the main workhorses of many biological activities. Small molecule drugs such as ibuprofen often work by inhibiting or promoting the function of proteins or their interactions with other biomolecules. Developing useful “virtual screening” methods where potential small molecules can be evaluated computationally rather than in a lab, has long been an area of research. However, the persistent challenge is to build a method that works well enough across a wide range of chemical space to be useful for finding small molecules with physically verified useful interaction with a protein of interest, i.e., “hits”.
In “Machine learning on DNA-encoded libraries: A new paradigm for hit-finding”, recently published in the Journal of Medicinal Chemistry, we worked in collaboration with X-Chem Pharmaceuticals to demonstrate an effective new method for finding biologically active molecules using a combination of physical screening with DNA-encoded small molecule libraries and virtual screening using a graph convolutional neural network (GCNN). This research has led to the creation of the Chemome initiative, a cooperative project between our Accelerated Science team and ZebiAI that will enable the discovery of many more small molecule chemical probes for biological research.
Background on Chemical Probes
Making sense of the biological networks that support life and produce disease is an immensely complex task. One approach to study these processes is using chemical probes, small molecules that aren’t necessarily useful as drugs, but that selectively inhibit or promote the function of specific proteins. When you have a biological system to study (such as cancer cells growing in a dish), you can add the chemical probe at a specific time and observe how the biological system responds differently when the targeted protein has increased or decreased activity. But, despite how useful chemical probes are for this kind of basic biomedical research, only 4% of human proteins have a known chemical probe available.
The process of finding chemical probes begins similarly to the earliest stages of small molecule drug discovery. Given a protein target of interest, the space of small molecules is scanned to find “hit” molecules that can be further tested. Robotic assisted high throughput screening where up to hundreds of thousands or millions of molecules are physically tested is a cornerstone of modern drug research. However, the number of small molecules you can easily purchase (1.2x109) is much larger than that, which in turn is much smaller than the number of small drug like molecules (estimates from 1020 to 1060). “Virtual screening” could possibly quickly and efficiently search this vast space of potentially synthesizable molecules and greatly speed up the discovery of therapeutic compounds.
DNA-Encoded Small Molecule Library Screening
The physical part of the screening process uses DNA-encoded small molecule libraries (DELs), which contain many distinct small molecules in one pool, each of which is attached to a fragment of DNA serving as a unique barcode for that molecule. While this basic technique has been around for several decades, the quality of the library and screening process is key to producing meaningful results.
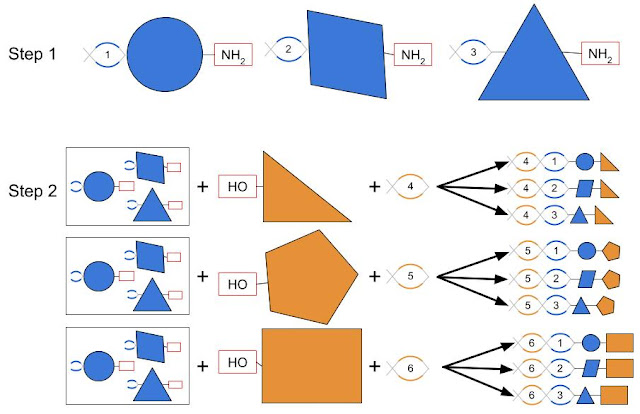
DELs are a very clever idea to solve a biochemical challenge, which is how to collect small molecules into one place with an easy way to identify each. The key is to use DNA as a barcode to identify each molecule, similar to Nobel Prize winning phage display technology. First, one generates many chemical fragments, each with a unique DNA barcode attached, along with a common chemical handle (the NH2 in this case). The results are then pooled and split into separate reactions where a set of distinct chemical fragments with another common chemical handle (e.g., OH) are added. The chemical fragments from the two steps react and fuse together at the common chemical handles. The DNA fragments are also connected to build one continuous barcode for each molecule. The net result is that by performing 2N operations, one gets N2 unique molecules, each of which is identified by its own unique DNA barcode. By using more fragments or more cycles, it’s relatively easy to make libraries with millions or even billions of distinct molecules.
 |
| An overview of the process of creating a DNA encoded small molecule library. First, DNA “barcodes” (represented here with numbered helices) are attached to small chemical fragments (the blue shapes) which expose a common chemical “handle” (e.g. the NH2 shown here). When mixed with other chemical fragments (the orange shapes) each of which has another exposed chemical “handle” (the OH) with attached DNA fragments, reactions merge the sets of chemical and DNA fragments, resulting in a voluminous library of small molecules of interest, each with a unique DNA “barcode”. |
Machine Learning on DEL Data
Given the physical screening data returned for a particular protein, we build an ML model to predict whether an arbitrarily chosen small molecule will bind to that protein. The physical screening with the DEL provides positive and negative examples for an ML classifier. To simplify slightly, the small molecules that remain at the end of the screening process are positive examples and everything else are negative examples. We use a graph convolutional neural network, which is a type of neural network specially designed for small graph-like inputs, such as the small molecules in which we are interested.
Results
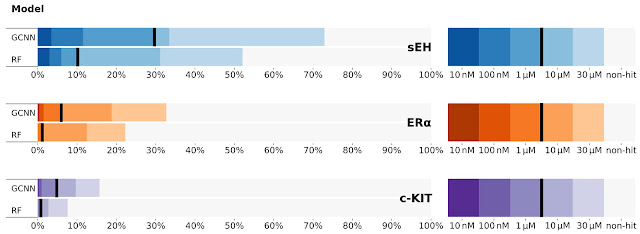
We physically screened three diverse proteins using DEL libraries: sEH (a hydrolase), ERα (a nuclear receptor), and c-KIT (a kinase). Using the DEL-trained models, we virtually screened large make-on-demand libraries from Mcule and an internal molecule library at X-Chem to identify a diverse set of molecules predicted to show affinity with each target. We compared the results of the GCNN models to a random forest (RF) model, a common method for virtual screening that uses standard chemical fingerprints, which we use as baseline. We find that the GCNN model significantly outperforms the RF model in discovering more potent candidates.
 |
| Fraction of molecules (“hit rates”) from those tested showing various levels of activity, comparing predictions from two different machine learned models (a GCNN and random forests, RF) on three distinct protein targets. The color scale on the right uses a common metric IC50 for representing the potency of a molecule. nM means “nanomolar” and µM means “micromolar”. Smaller values / darker colors are generally better molecules. Note that typical virtual screening approaches not built with DEL data normally only reach a few percent on this scale. |
The Chemome Initiative
ZebiAI Therapeutics was founded based on the results of this research and has partnered with our team and X-Chem Pharmaceuticals to apply these techniques to efficiently deliver new chemical probes to the research community for human proteins of interest, an effort called the Chemome Initiative.
As part of the Chemome Initiative, ZebiAI will work with researchers to identify proteins of interest and source screening data, which our team will use to build machine learning models and make predictions on commercially available libraries of small molecules. ZebiAI will provide the predicted molecules to researchers for activity testing and will collaborate with researchers to advance some programs through discovery. Participation in the program requires that the validated hits be published within a reasonable time frame so that the whole community can benefit. While more validation must be done to make the hit molecules useful as chemical probes, especially for specifically targeting the protein of interest and the ability to function correctly in common assays, having potent hits is a big step forward in the process.
We’re excited to be a part of the Chemome Initiative enabled by the effective ML techniques described here and look forward to its discovery of many new chemical probes. We expect the Chemome will spur significant new biological discoveries and ultimately accelerate new therapeutic discovery for the world.
Acknowledgements
This work represents a multi-year effort between the Accelerated Science Team and X-Chem Pharmaceuticals with many people involved. This project would not have worked without the combined diverse skills of biologists, chemists, and ML researchers. We should especially acknowledge Eric Sigel (of X-Chem, now at ZebiAI) and Kevin McCloskey (of Google), the first authors on the paper and Steve Kearnes (of Google) for core modelling ideas and technical work.
Quick links
Other posts of interest
-

April 9, 2026
ConvApparel: Measuring and bridging the realism gap in user simulators- Generative AI ·
- Machine Intelligence ·
- Natural Language Processing
-

April 3, 2026
Evaluating alignment of behavioral dispositions in LLMs- Generative AI ·
- Human-Computer Interaction and Visualization ·
- Machine Intelligence
-

March 31, 2026
Building better AI benchmarks: How many raters are enough?- Algorithms & Theory ·
- Machine Intelligence
×
❮
❯