
Smarter nucleic acid design with NucleoBench and AdaBeam
September 11, 2025
Cory Y. McLean, Senior Staff Software Engineer, Google Research
We developed an open-source software benchmark for nucleic acid sequence design, and introduced a novel algorithm, AdaBeam, that outperforms existing algorithms on 11 the 16 tasks, demonstrating superior scaling properties on long sequences and large predictors.
Quick links
Designing new DNA and RNA sequences with specific therapeutic properties is a critical challenge in modern medicine. These molecules are the building blocks for next-generation treatments, from more precise CRISPR gene therapies to more stable and effective mRNA vaccines. However, finding the right sequence is like searching for a single grain of sand on a vast beach. For instance, a small functional region of an RNA molecule called the 5' UTR can be one of over 2x10120 possible sequences, making a brute-force search to optimize its function impossible.
What if we could use AI to navigate this vast search space, drastically cutting down the time and cost of drug discovery? While various efforts have made great strides in developing AI models that predict the properties of a given nucleic acid sequence, there remains opportunity to innovate on the algorithms that use these models to generate optimal sequences. A lack of standardized evaluation hinders progress and prevents us from translating powerful predictive models into the best possible therapeutic molecules.
To address this gap, in a research collaboration between Google Research and Move37 Labs, we introduce NucleoBench, the first large-scale, standardized benchmark for comparing nucleic acid design algorithms. By running over 400,000 experiments across 16 distinct biological challenges, we've created a framework to rigorously evaluate and understand how different algorithms perform. The insights from this work enabled us to develop AdaBeam, a hybrid design algorithm that outperforms existing methods on 11 of the 16 tasks and scales more effectively to the large and complex models that are defining the future of AI in biology. We have made AdaBeam and all of our algorithm implementations freely available to spur further innovation.
The core challenge
The process of designing a new nucleic acid sequence using computers generally follows four steps:
- Generate data: Collect a high-quality dataset of sequences with the desired property (e.g., binding to a cancer-related protein).
- Train a predictive model: Use this data to train a model (often a neural network) that can predict the property from a DNA or RNA sequence.
- Generate candidate sequences: This is the crucial design step. Use an optimization algorithm to generate new sequences that the model predicts will have the highest possible score for the desired property.
- Validate candidates: Synthesize and test the most promising sequences in a wet lab to see if they work as predicted.
- Retrain [Optional]: Retrain the model on validation data.
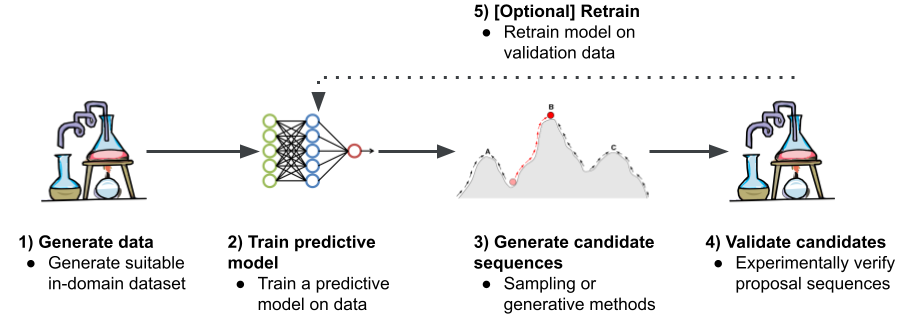
The typical workflow for computational nucleic acid design.
In this work we focus on the design algorithms of step 3. At present, different research groups use different algorithms and test them on different tasks, making it impossible to know which methods are truly the best. Most existing benchmarks rely on algorithms like simulated annealing or vanilla genetic algorithms, which were developed decades before modern deep learning and cannot take advantage of crucial information, like gradients, from the neural network models.
NucleoBench
To create a comprehensive and fair benchmark, we selected a diverse slate of gradient-free and gradient-based design algorithms. Gradient-free algorithms include well-established methods like directed evolution and simulated annealing, which are inspired by processes in evolution and physics, respectively. These algorithms treat the predictive AI model as a "black box", and test new sequences without needing to understand how the model works internally. Their strength lies in their simplicity and broad applicability, but this means they potentially miss out on valuable clues from the model.
Gradient-based design algorithms leverage the internal workings of neural networks and include more modern algorithms like FastSeqProp and Ledidi. They use the model's gradients (i.e., the direction of steepest improvement) to intelligently guide the search for better sequences, but take longer to compute than just using the output of the neural network.
To our knowledge, NucleoBench is the most comprehensive benchmark for nucleic acid design algorithms thus far and allows for a fair, apples-to-apples comparison between algorithms. We evaluated 9 different algorithms on the same 16 tasks with the same starting sequences, giving us unprecedented statistical power to draw meaningful conclusions. These tasks span a wide range of biological challenges, including:
- Controlling gene expression in specific cell types (e.g., liver or neuronal cells)
- Maximizing the binding of transcription factors (proteins that regulate genes)
- Improving the physical accessibility of chromatin for biomolecular interactions
- Predicting gene expression from very long DNA sequences using large-scale models like Enformer
| Task Category | Description | Num Tasks | Seq Len (bp) | Speed (ms / example) |
| Cell-type specific cis-regulatory activity | How DNA sequences control gene expression from the same DNA molecule. Cell types include: precursor blood cells, liver cells, neuronal cells | 3 | 200 | 2 |
| Transcription factor binding | How likely a specific transcription factor will bind to a particular stretch of DNA | 11 | 3000 | 55 |
| Chromatin accessibility | How physically accessible DNA is for interactions with other molecules | 1 | 3000 | 260 |
| Selective gene expression | Prediction of gene expression | 1 | 196,608 / 256* | 15,000 |
Summary of design tasks in NucleoBench. *Model input length is 200K base pairs (bp), but only 256 bp are edited.
We introduced ordered and unordered beam search algorithms, staples from computer science, to test how fixing the order of sequence edits compares to a more flexible, random-order approach. We also created Gradient Evo, a novel hybrid that enhances the directed evolution algorithm by using model gradients to guide its mutations to independently evaluate how important gradients were for edit location selection versus selecting a specific edit.
We also developed AdaBeam, a hybrid adaptive beam search algorithm that combines the most effective elements of unordered beam search with AdaLead, a top-performing, non-gradient design algorithm. Adaptive search algorithms don't typically explore randomly; instead, their behavior changes as a result of the search to focus their efforts on the most promising areas of the sequence space. AdaBeam’s hybrid approach maintains a "beam", or a collection of the best candidate sequences found so far, and greedily expands on particularly promising candidates until they’ve been sufficiently explored.
In practice, AdaBeam begins with a population of candidate sequences and their scores. In each round, it first selects a small group of the highest-scoring sequences to act as "parents". For each parent, AdaBeam generates a new set of "child" sequences by making a random number of random-but-guided mutations. It then follows a short, greedy exploration path, allowing the algorithm to quickly "walk uphill" in the fitness landscape. After sufficient exploration, all the newly generated children are pooled together, and the algorithm selects the absolute best ones to form the starting population for the next round, repeating the cycle. This process of adaptive selection and targeted mutation allows AdaBeam to efficiently focus on high-performing sequences.
Computer-assisted design tasks pose difficult engineering problems, owing to the incredibly large search space. These difficulties become more acute as we attempt to design longer sequences, such as mRNA sequences, and use modern, large neural networks to guide the design. AdaBeam is particularly efficient on long sequences by using fixed-compute probabilistic sampling instead of computations that scale with sequence length. To enable AdaBeam to work with large models, we reduce peak memory consumption during design by introducing a trick we call “gradient concatenation.” However, existing design algorithms that don’t have these features have difficulty scaling to long sequences and large models. Gradient-based algorithms are particularly affected. To facilitate a fair comparison, we limit the length of the designed sequences, even though AdaBeam can scale longer and larger. For example, even though the DNA expression prediction model Enformer runs on ~200K nucleotide sequences, we limit design to just 256 nucleotides.

Summary of design algorithms in NucleoBench. Below the solid line are design algorithms devised in this work.
Evaluation
We evaluate each design algorithm based on the final fitness score of the sequence each produced. The fitness score is defined as how well the sequence performed on the biological task according to the predictive model. To ensure fairness, we ran over 400,000 experiments where each design algorithm was given a fixed amount of time and the exact same 100 starting sequences for each task. We also measured the convergence speed, tracking how quickly each algorithm found its best solution, as faster algorithms save valuable time and computational resources.
We characterized performance variability by measuring how much an algorithm's final score was influenced by random chance versus its starting sequence. We quantified the effect of algorithmic randomness by re-running experiments with five different random seeds. To assess the impact of the starting point, we analyzed the variance in final scores across the 100 identical start sequences given to each design algorithm. We used a Friedman test to investigate whether "intrinsically difficult start sequences", or sequences that are hard for all algorithms to optimize, exist.
To assess the distribution of performance ranks, we compared the final performance for each of the nine algorithms across every experiment in the NucleoBench benchmark for each unique combination of a task and a starting sequence. A rank-based "order score" from 0 to 8 was then assigned, with 0 going to the best-performing algorithm, 1 to the second-best, and so on. Each violin shape is constructed by aggregating all the rank scores a single algorithm received across the 400,000+ experiments, with the width of the violin at any point showing how frequently that algorithm achieved a particular rank.
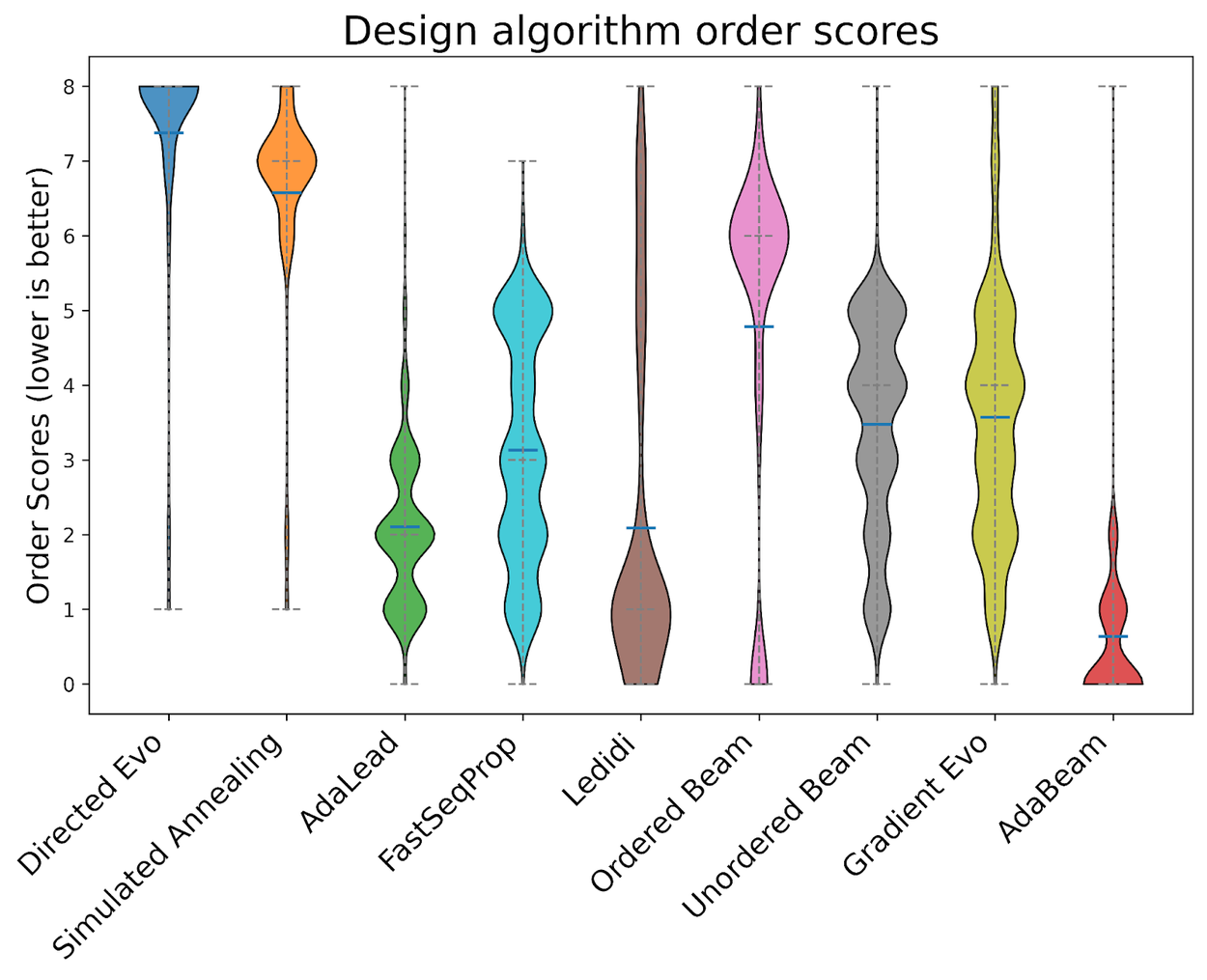
The distribution of final scores for each algorithm. X-axis is the design algorithm, y-axis is the aggregate order score. Order scores are determined by assigning an integer [0, 9] for each (task, start sequence, design algorithm) tuple according to the performances of all the final sequences for that (task, start sequence) pair. 0 is the top performer. Aggregate scores are computed by averaging over all such scores.
Gradient-based methods were the reigning champions amongst existing methods. However, we found that AdaBeam outperformed them, demonstrating that relying on gradients is not the only path to top-tier performance and scalability.
AdaBeam improves upon previous methods in several key ways:
- Efficiency: It replaces AdaLead’s sampling step with a faster calculation, doubling its speed on long sequences.
- Smart Exploration: It uses a significantly more effective "unordered" approach to deciding where to edit a sequence.
- Advanced Engineering: It uses gradient concatenation to substantially reduce memory usage, enabling application to massive models like Enformer.
Across the 16 tasks in NucleoBench, AdaBeam was the best-performing algorithm 11 times. It also proved to be one of the fastest to converge on a high-quality solution, demonstrating superior scaling properties that are essential for tackling the next generation of AI challenges in biology.
Future directions
Our NucleoBench benchmark reveals the importance of rigorous, standardized evaluation and uncovers surprising findings, such as the critical impact of the initial sequence and the ineffectiveness of some established algorithm features. However, significant challenges remain. The best gradient-based methods still struggle to scale to the largest models and longest sequences, and substantial scalability gains can be realized through better software engineering. While our new algorithm, AdaBeam, sets a new state-of-the-art, future work must focus on algorithms that adhere to biological constraints and improve scalability.
A core principle of our work is a commitment to biosafety and responsible innovation. While AdaBeam represents a step forward for biological sequence design, it only improves the optimization according to a pre-existing predictive model. In other words, it is an optimizer, not an originator; the algorithm can only design sequences to maximize a goal defined by a user-provided predictive model. By releasing AdaBeam as an open-source tool, we empower researchers while ensuring the “human-in-the-loop” remains central to the design of biological molecules. Algorithms like AdaBeam can help scientists design more effective mRNA vaccines, create safer CRISPR gene therapies, and develop novel treatments for a wide range of diseases, bringing the promise of AI-driven drug discovery closer to reality.
Acknowledgments
This work represents a collaboration between Joel Shor (Move37 Labs), Erik Strand (Move37 Labs, MIT), and Cory Y. McLean (Google Research). We thank Sager Gosai, Daniel Friedman, Anna Lewis, Vikram Agarwal, and Michael Brenner for their guidance, discussions, and support throughout this project.
Quick links
Other posts of interest
-

April 3, 2026
Evaluating alignment of behavioral dispositions in LLMs- Generative AI ·
- Human-Computer Interaction and Visualization ·
- Machine Intelligence
-

March 31, 2026
Building better AI benchmarks: How many raters are enough?- Algorithms & Theory ·
- Machine Intelligence
-

March 25, 2026
Vibe Coding XR: Accelerating AI + XR prototyping with XR Blocks and Gemini- Human-Computer Interaction and Visualization ·
- Machine Intelligence

