Improving Genomic Discovery with Machine Learning
June 23, 2021
Posted by Andrew Carroll, Product Manager and Cory McLean, Software Engineer, Google Health
Quick links
Each person’s genome, which collectively encodes the biochemical machinery they are born with, is composed of over 3 billion letters of DNA. However, only a small subset of the genome (~4-5 million positions) varies between two people. Nonetheless, each person’s unique genome interacts with the environment they experience to determine the majority of their health outcomes. A key method of understanding the relationship between genetic variants and traits is a genome-wide association study (GWAS), in which each genetic variant present in a cohort is individually examined for correlation with the trait of interest. GWAS results can be used to identify and prioritize potential therapeutic targets by identifying genes that are strongly associated with a disease of interest, and can also be used to build a polygenic risk score (PRS) to predict disease predisposition based on the combined influence of variants present in an individual. However, while accurate measurement of traits in an individual (called phenotyping) is essential to GWAS, it often requires painstaking expert curation and/or subjective judgment calls.
In “Large-scale machine learning-based phenotyping significantly improves genomic discovery for optic nerve head morphology”, we demonstrate how using machine learning (ML) models to classify medical imaging data can be used to improve GWAS. We describe how models can be trained for phenotypes to generate trait predictions and how these predictions are used to identify novel genetic associations. We then show that the novel associations discovered improve PRS accuracy and, using glaucoma as an example, that the improvements for anatomical eye traits relate to human disease. We have released the model training code and detailed documentation for its use on our Genomics Research GitHub repository.
Identifying genetic variants associated with eye anatomical traits
Previous work has demonstrated that ML models can identify eye diseases, skin diseases, and abnormal mammogram results with accuracy approaching or exceeding state-of-the-art methods by domain experts. Because identifying disease is a subset of phenotyping, we reasoned that ML models could be broadly used to improve the speed and quality of phenotyping for GWAS.
To test this, we chose a model that uses a fundus image of the eye to accurately predict whether a patient should be referred for assessment for glaucoma. This model uses the fundus images to predict the diameters of the optic disc (the region where the optic nerve connects to the retina) and the optic cup (a whitish region in the center of the optic disc). The ratio of the diameters of these two anatomical features (called the vertical cup-to-disc ratio, or VCDR) correlates strongly with glaucoma risk.
 |
| A representative retinal fundus image showing the vertical cup-to-disc ratio, which is an important diagnostic measurement for glaucoma. |
We applied this model to predict VCDR in all fundus images from individuals in the UK Biobank, which is the world’s largest dataset available to researchers worldwide for health-related research in the public interest, containing extensive phenotyping and genetic data for ~500,000 pseudonymized (the UK Biobank's standard for de-identification) individuals. We then performed GWAS in this dataset to identify genetic variants that are associated with the model-based predictions of VCDR.
 |
| Applying a VCDR prediction model trained on clinical data to generate predicted values for VCDR to enable discovery of genetic associations for the VCDR trait. |
The ML-based GWAS identified 156 distinct genomic regions associated with VCDR. We compared these results to a VCDR GWAS conducted by another group on the same UK Biobank data, Craig et al. 2020, where experts had painstakingly labeled all images for VCDR. The ML-based GWAS replicates 62 of the 65 associations found in Craig et al., which indicates that the model accurately predicts VCDR in the UK Biobank images. Additionally, the ML-based GWAS discovered 93 novel associations.
 |
| Number of statistically significant GWAS associations discovered by exhaustive expert labeling approach (Craig et al., left), and by our ML-based approach (right), with shared associations in the middle. |
The ML-based GWAS improves polygenic model predictions
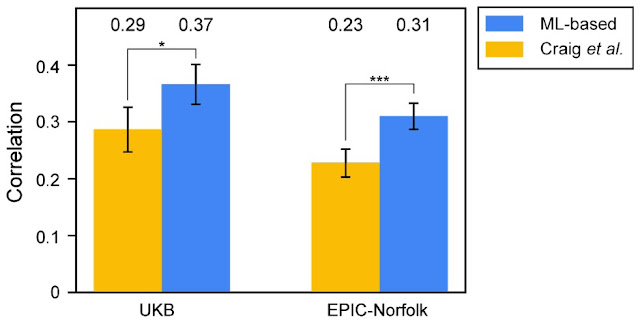
To validate that the novel associations discovered in the ML-based GWAS are biologically relevant, we developed independent PRSes using the Craig et al. and ML-based GWAS results, and tested their ability to predict human-expert-labeled VCDR in a subset of UK Biobank as well as a fully independent cohort (EPIC-Norfolk). The PRS developed from the ML-based GWAS showed greater predictive ability than the PRS built from the expert labeling approach in both datasets, providing strong evidence that the novel associations discovered by the ML-based method influence VCDR biology, and suggesting that the improved phenotyping accuracy (i.e., more accurate VCDR measurement) of the model translates into a more powerful GWAS.
 |
| The correlation between a polygenic risk score (PRS) for VCDR generated from the ML-based approach and the exhaustive expert labeling approach (Craig et al.). In these plots, higher values on the y-axis indicate a greater correlation and therefore greater prediction from only the genetic data. [* — p ≤ 0.05; *** — p ≤ 0.001] |
As a second validation, because we know that VCDR is strongly correlated with glaucoma, we also investigated whether the ML-based PRS was correlated with individuals who had either self-reported that they had glaucoma or had medical procedure codes suggestive of glaucoma or glaucoma treatment. We found that the PRS for VCDR determined using our model predictions were also predictive of the probability that an individual had indications of glaucoma. Individuals with a PRS 2.5 or more standard deviations higher than the mean were more than 3 times as likely to have glaucoma in this cohort. We also observed that the VCDR PRS from ML-based phenotypes was more predictive of glaucoma than the VCDR PRS produced from the extensive manual phenotyping.
 |
| The odds ratio of glaucoma (self-report or ICD code) stratified by the PRS for VCDR determined using the ML-based phenotypes (in standard deviations from the mean). In this plot, the y-axis shows the probability that the individual has glaucoma relative to the baseline rate (represented by the dashed line). The x-axis shows standard deviations from the mean for the PRS. Data are visualized as a standard box plot, which illustrates values for the mean (the orange line), first and third quartiles, and minimum and maximum. |
Conclusion
We have shown that ML models can be used to quickly phenotype large cohorts for GWAS, and that these models can increase statistical power in such studies. Although these examples were shown for eye traits predicted from retinal imaging, we look forward to exploring how this concept could generally apply to other diseases and data types.
Acknowledgments
We would like to especially thank co-author Dr. Anthony Khawaja of Moorfields Eye Hospital for contributing his extensive medical expertise. We also recognize the efforts of Professor Jamie Craig and colleagues for their exhaustive labeling of UK Biobank images, which allowed us to make comparisons with our method. Several authors of that work, as well as Professor Stuart MacGregor and collaborators in Australia and at Max Kelsen have independently replicated these findings, and we value these scientific contributions as well. Last, this work summarizes the work of the following Google contributors, who we would like to thank: Babak Alipanahi, Farhad Hormozdiari, Babak Behsaz, Justin Cosentino, Zachary R. McCaw, Emanuel Schorsch, D. Sculley, Elizabeth H. Dorfman, Sonia Phene, Naama Hammel, Andrew Carroll, and Cory Y. McLean
Quick links
Other posts of interest
-

March 31, 2026
Building better AI benchmarks: How many raters are enough?- Algorithms & Theory ·
- Machine Intelligence
-

March 25, 2026
Vibe Coding XR: Accelerating AI + XR prototyping with XR Blocks and Gemini- Human-Computer Interaction and Visualization ·
- Machine Intelligence
-

March 24, 2026
TurboQuant: Redefining AI efficiency with extreme compression- Algorithms & Theory ·
- Generative AI ·
- Machine Intelligence