
Highly accurate genome polishing with DeepPolisher: Enhancing the foundation of genomic research
August 6, 2025
Kishwar Shafin, Technical Lead, and Andrew Carroll, Product Lead, Google Research
DeepPolisher, is a new deep learning tool that significantly improves the accuracy of genome assemblies by precisely correcting base-level errors, which recently played a key role in enhancing the Human Pangenome Reference.
The key to understanding heredity, disease, and evolution lies in the genome, which is encoded in nucleotides (i.e., the bases A, T, G, and C). DNA sequencers can read these nucleotides, but doing so both accurately and at scale is challenging, due to the very small scale of the base pairs. However, to unlock the secrets hidden within the genome, we must be able to assemble a reference genome as close to perfect as possible.
Errors in assembly can limit the methods used to identify genes and proteins, and can cause later diagnostic processes to miss disease-causing variants. In genome assembly, the same genome is sequenced many times, allowing iterative correction of errors. Still, with the human genome being 3 billion nucleotides, even a small error rate can mean a large total number of errors and can limit the derived genome’s utility.
In an effort to continually improve the resources for genome assembly, we introduce DeepPolisher, an open-source method for genome assembly that we developed in a collaboration with the UC Santa Cruz Genomics Institute. In our recent paper, “Highly accurate assembly polishing with DeepPolisher”, published in Genome Research, we describe how this pipeline extends existing methods to improve the accuracy of the genome assembly. DeepPolisher reduces the number of errors in the assembly by 50% and the number of insertion or deletion (“indel”) errors by 70%. This is especially important since indel errors interfere with the identification of genes.
Background
While there are several ways to measure DNA, most typically involve capturing the process of copying DNA. One method for this involves attaching label molecules with different colors to separate building block nucleotides and observing the process of each being added to the DNA molecule being copied. The DNA copying machinery always copies the strand in a particular orientation, so although the information is redundantly encoded on both strands, only nucleotides from one strand are read at a time. Identifying the nucleotides requires detectors that are able to resolve single molecules, which limits the accuracy of measurements.
One breakthrough technology to scale this method, developed by Illumina, copies one molecule of the DNA to be sequenced into a cluster of identical copies. It then monitors as the cluster copies in sync, thus increasing the signal for each base. However, as it is impossible to ensure the cluster copies in perfect unison, the cluster may desynchronize so that the signal of different bases blend together, which limits the lengths of the DNA measured using this method to a few hundred nucleotides.
Although these sequences (called “reads”) are short, they are still useful for analysis. By comparing them to a reference genome, i.e., an existing map of the genome of the species to be sequenced, it is possible to map many of the short reads to that reference, thus building up a more complete genome of the sampled individual. This can then be compared to the reference to better understand how the subject’s genome varies.

The human genome is composed of two strands that redundantly encode information (left), organized into chromosomes, with one full copy inherited from each parent (right). (Images from NHGRI)
Even with improved sequencing technology, there remain several challenges. First, the method relies on having a robust reference genome, which is itself exceptionally difficult to create. Even with such a reference, some parts of the genome look more like other parts, making them difficult to confidently map to the reference.
To address those challenges, scientists developed processes that could sequence individual molecules, enabling reads of tens of thousands of nucleotides. Initially, this process had unacceptable error rates (~10%). This was addressed when Pacific Biosciences developed a way to sequence the same molecule in multiple passes, reducing the error rate to only 1%, similar to the short-read methods. Google and Pacific Biosciences worked together on the first demonstration of this on a human genome.
Our team then took this further by developing DeepConsensus, which uses a sequence transformer to more accurately construct the correct sequence from the initial error-prone bases. Today Pacific Biosciences deploys DeepConsensus on their long-read sequencers to reduce the error rate to less than 0.1%. While this error rate is markedly better than the prior state of the art, reaching the accuracy required to construct a new, nearly perfect reference genome, requires combining sequence reads from multiple DNA molecules of the same individual to further correct remaining errors.
DeepPolisher
This is where DeepPolisher comes in. Adapted from DeepConsensus, DeepPolisher uses a Transformer architecture trained on the genome from a human cell line donated to the Personal Genomes Project. This reference genome has been exhaustively characterized by NIST and NHGRI and sequenced using many different technologies. It is estimated to be ~100% complete with a correctness of 99.99999%. This corresponds to around 300–1000 total errors across the 6 billion nucleotides in the genome (two copies of the 3 billion nucleotide reference inherited from each parent).
By conducting PacBio sequencing and genome assembly, we can identify remaining errors and then train models to learn to correct them. For training, the model takes the sequenced bases, their quality, and how uniquely they map to a given part of the reference assembly. During training, we use only chromosomes 1–19. We hold out chromosomes 20–22, using the performance on chromosomes 21 and 22 to select a model, and we report accuracies using chromosome 20.
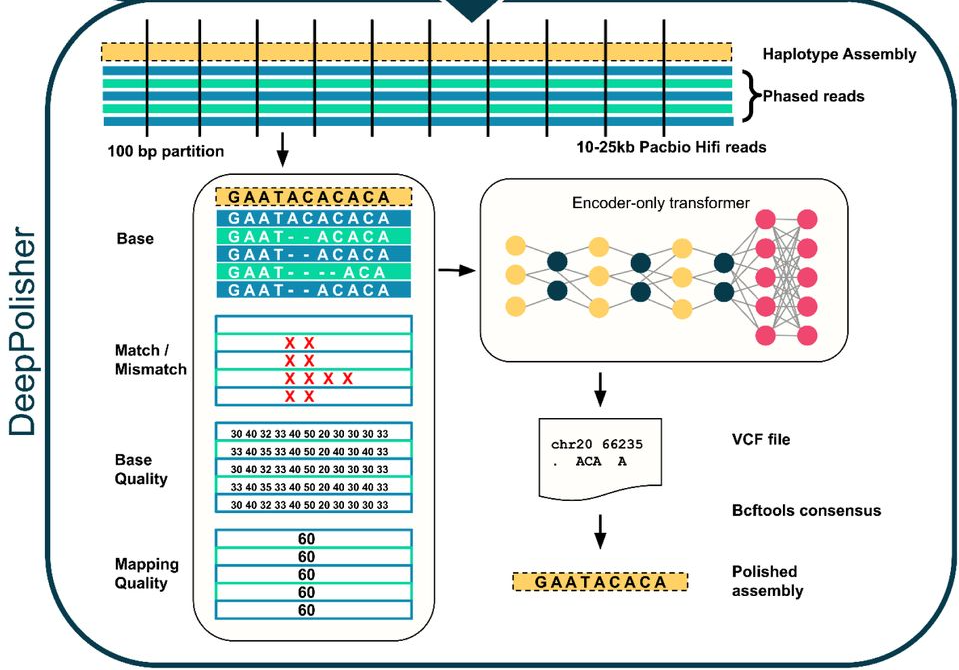
Architecture of DeepPolisher. The sequence reads are categorized by parental origin (called “phasing”) and are aligned to the draft genome assembly. The input channels are: the base information, reported quality by the sequencer, the quality of the mapping (ability to place the reads uniquely on the assembly), and annotations of mismatched bases. This is sent to an encoder-only Transformer, which classifies the errors in the assembly and then suggests a fix, which is used to correct the assembly.
Performance
DeepPolisher reduces errors in a genome assembly by approximately half, an improvement largely driven by the reduction in insertion–deletion (“indel”) errors, which decrease by more than 70 percent. Reducing these types of errors is especially important, because inserted or deleted bases can shift the reading frame of a gene, causing annotation programs to overlook that gene when labelling the genome and hiding it from reports in clinical analysis or drug discovery.
We quantify the quality of a genome using a “Q-score”, which is a base-10 logarithm of the probability that a position in the genome has an error. A Q30 score means 99.9% chance of being correct, while a Q60 means a 99.9999% chance of a base being correct. To assess the improvement of DeepPolisher, we pulled sequencing data being used to assemble new genomes for the Human Pangenome Reference Consortium (HPRC). We looked for potential errors in the assembly by trying to identify combinations of nucleotides in the assembly that don’t occur in other sequencing of the same sample with different sequencing technologies. By doing this analysis in the parts of the genome for which the other sequencing method has no systematic biases (confident region), we can show an improvement of the assembly from Q66.7 to Q70.1 on average. We also show improvement in every single sample assessed.
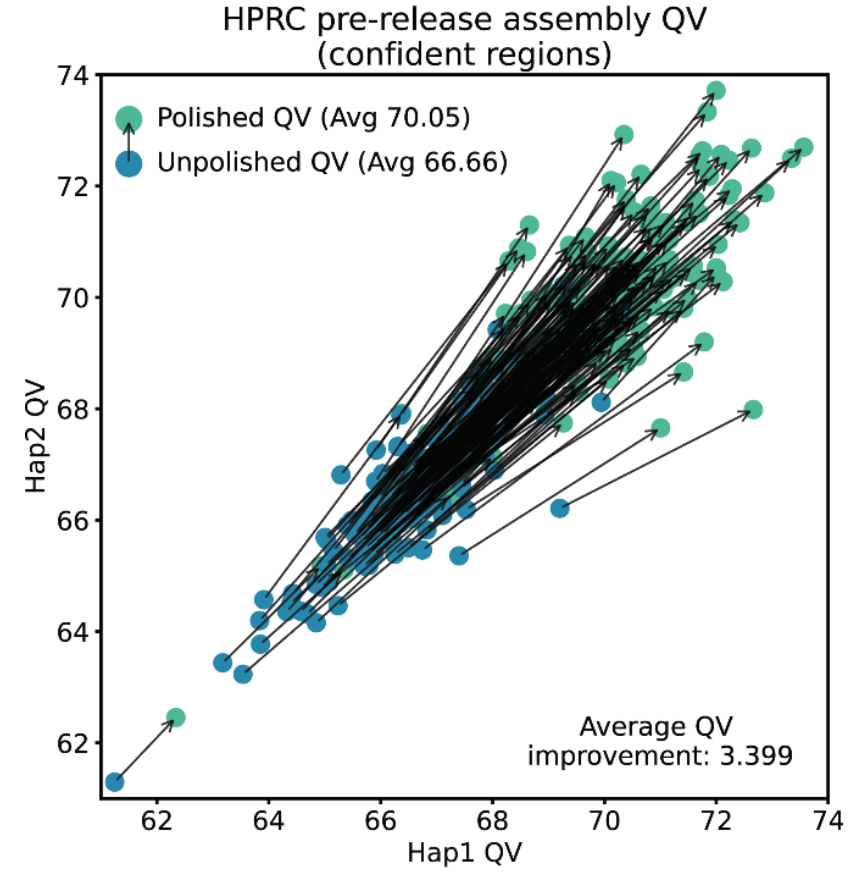
Assembly qualities before and after polishing for 180 samples. For each sample, the genome is separated by the parental origin (the copy of the genome transmitted by father or mother) indicated as Haplotype (Hap) 1 or 2, and the assessed quality of those haplotypes.
Deployment
DeepPolisher is already being used to improve genomics resources for the scientific community. In May, the HPRC announced their second data release, which included sequenced genome assemblies on 232 individuals, a fivefold increase over the first release. The data in the second release underwent an additional polishing step with DeepPolisher that reduced single nucleotide and indel errors twofold, leading to an extremely low error rate of less than one base error in half a million assembled bases.
By providing DeepPolisher as an open-source tool, our goal is to make the methods available broadly to the community. Working with the Human Pangenome Reference Consortium, we help enable scientists to more accurately diagnose genetic diseases for individuals of all ancestries.
Acknowledgements
This blog post demonstrates Google’s contribution to the development of DeepPolisher for improving the quality of genome assemblies. Integrating DeepPolisher in the broader context of generating highly accurate pangenome references involves contributions from nearly 195 authors from 68 different organizations. We thank the research groups from UCSC Genomics Institute (GI) under Professor Benedict Paten and Professor Karen Miga for helping in primary analysis and development directions of DeepPolisher. We acknowledge Mira Mastoras and Mobin Asri for leading the core analysis and integration of DeepPolisher to the pangenome generation pipeline. We thank the Google technical contributors: Pi-Chuan Chang, Daniel E. Cook, Alexey Kolesnikov, Lucas Brambrink, and Maria Nattestad. We thank Lizzie Dorfman, Dale Webster, and Katherine Chou for strategic leadership, and Monique Brouillette for help in writing.
-
Labels:
- General Science
- Health & Bioscience
Other posts of interest
-

March 17, 2026
Google Research at The Check Up: from healthcare innovation to real-world care settings- Health & Bioscience ·
- Machine Intelligence
-
March 17, 2026
Improving breast cancer screening workflows with machine learning- Health & Bioscience ·
- Human-Computer Interaction and Visualization
-

March 16, 2026
Testing LLMs on superconductivity research questions- Education Innovation ·
- General Science ·
- Machine Intelligence ·
- Natural Language Processing

