DeepVariant Accuracy Improvements for Genetic Datatypes
April 19, 2018
Posted by Pi-Chuan Chang, Software Engineer and Lizzie Dorfman, Technical Program Manager, Google Brain Team
Quick links
Last December we released DeepVariant, a deep learning model that has been trained to analyze genetic sequences and accurately identify the differences, known as variants, that make us all unique. Our initial post focused on how DeepVariant approaches “variant calling” as an image classification problem, and is able to achieve greater accuracy than previous methods.
Today we are pleased to announce the launch of DeepVariant v0.6, which includes some major accuracy improvements. In this post we describe how we train DeepVariant, and how we were able to improve DeepVariant's accuracy for two common sequencing scenarios, whole exome sequencing and polymerase chain reaction sequencing, simply by adding representative data into DeepVariant's training process.
Many Types of Sequencing Data
Approaches to genomic sequencing vary depending on the type of DNA sample (e.g., from blood or saliva), how the DNA was processed (e.g., amplification techniques), which technology was used to sequence the data (e.g., instruments can vary even within the same manufacturer) and what section or how much of the genome was sequenced. These differences result in a very large number of sequencing "datatypes".
Typically, variant calling tools have been tuned for one specific datatype and perform relatively poorly on others. Given the extensive time and expertise involved in tuning variant callers for new datatypes, it seemed infeasible to customize each tool for every one. In contrast, with DeepVariant we are able to improve accuracy for new datatypes simply by including representative data in the training process, without negatively impacting overall performance.
Truth Sets for Variant Calling
Deep learning models depend on having high quality data for training and evaluation. In the field of genomics, the Genome in a Bottle (GIAB) consortium, which is hosted by the National Institute of Standards and Technology (NIST), produces human genomes for use in technology development, evaluation, and optimization. The benefit of working with GIAB benchmarking genomes is that their true sequence is known (at least to the extent currently possible). To achieve this, GIAB takes a single person's DNA and repeatedly sequences it using a wide variety of laboratory methods and sequencing technologies (i.e. many datatypes) and analyzes the resulting data using many different variant calling tools. A tremendous amount of work then follows to evaluate and adjudicate discrepancies to produce a high-confidence "truth set" for each genome.
The majority of DeepVariant’s training data is from the first benchmarking genome released by GIAB, HG001. The sample, from a woman of northern European ancestry, was made available as part of the International HapMap Project, the first large-scale effort to identify common patterns of human genetic variation. Because DNA from HG001 is commercially available and so well characterized, it is often the first sample used to test new sequencing technologies and variant calling tools. By using many replicates and different datatypes of HG001, we can generate millions of training examples which helps DeepVariant learn to accurately classify many datatypes, and even generalize to datatypes it has never seen before.
Improved Exome Model in v0.5
In the v0.5 release we formalized a benchmarking-compatible training strategy to withhold from training a complete sample, HG002, as well as any data from chromosome 20. HG002, the second benchmarking genome released by GIAB, is from a male of Ashkenazi Jewish ancestry. Testing on this sample, which differs in both sex and ethnicity from HG001, helps to ensure that DeepVariant is performing well for diverse populations. Additionally reserving chromosome 20 for testing guarantees that we can evaluate DeepVariant's accuracy for any datatype that has truth data available.
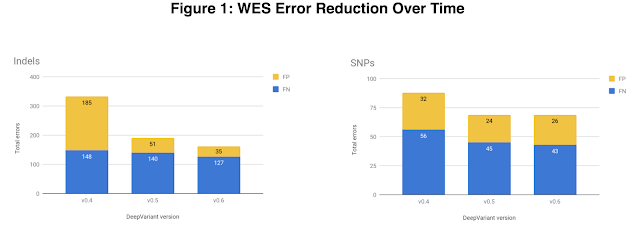
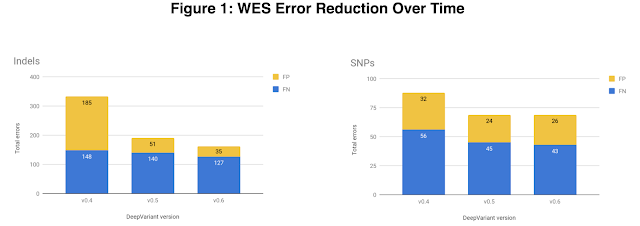
In v0.5 we also focused on exome data, which is the subset of the genome that directly codes for proteins. The exome is only ~1% of the whole human genome, so whole exome sequencing (WES) costs less than whole genome sequencing (WGS). The exome also harbors many variants of clinical significance which makes it useful for both researchers and clinicians. To increase exome accuracy we added a variety of WES datatypes, provided by DNAnexus, to DeepVariant's training data. The v0.5 WES model shows 43% fewer indel (insertion-deletion) errors and a 22% reduction in single nucleotide polymorphism (SNP) errors.
 |
| The total number of exome errors for HG002 across DeepVariant versions, broken down by indel errors (left) and SNP errors (right). Errors are either false positive (FP), colored yellow, or false negative (FN), colored blue. The largest accuracy jump is between v0.4 and v0.5, largely attributable to a reduction in indel FPs. |
Our newest release of DeepVariant, v0.6, focuses on improved accuracy for data that has undergone DNA amplification via polymerase chain reaction (PCR) prior to sequencing. PCR is an easy and inexpensive way to amplify very small quantities of DNA, and once sequenced results in what is known as PCR positive (PCR+) sequencing data. It is well known, however, that PCR can be prone to bias and errors, and non-PCR-based (or PCR-free) DNA preparation methods are increasingly common. DeepVariant's training data prior to the v0.6 release was exclusively PCR-free data, and PCR+ was one of the few datatypes for which DeepVariant had underperformed in external evaluations. By adding PCR+ examples to DeepVariant's training data, also provided by DNAnexus, we have seen significant accuracy improvements for this datatype, including a 60% reduction in indel errors.
 |
| DeepVariant v0.6 shows major accuracy improvements for PCR+ data, largely attributable to a reduction in indel errors. Here we re-analyze two PCR+ samples that were used in external evaluations, including DNAnexus on the left (see details in figure 10) and bcbio on the right, showing how indel accuracy improves with each DeepVariant version. |
Looking Forward
We released DeepVariant as open source software to encourage collaboration and to accelerate the use of this technology to solve real world problems. As the pace of innovation in sequencing technologies continues to grow, including more clinical applications, we are optimistic that DeepVariant can be further extended to produce consistent and highly accurate results. We hope that researchers will use DeepVariant v0.6 to accelerate discoveries, and if there is a sequencing datatype that you would like to see us prioritize, please let us know.
Quick links
Other posts of interest
-

March 31, 2026
Building better AI benchmarks: How many raters are enough?- Algorithms & Theory ·
- Machine Intelligence
-

March 25, 2026
Vibe Coding XR: Accelerating AI + XR prototyping with XR Blocks and Gemini- Human-Computer Interaction and Visualization ·
- Machine Intelligence
-

March 24, 2026
TurboQuant: Redefining AI efficiency with extreme compression- Algorithms & Theory ·
- Generative AI ·
- Machine Intelligence
×
❮
❯